Tres millones de personas en España, 30 millones en Europa y 300 millones en todo el mundo conviven con una enfermedad rara. En la actualidad, son más de 7.000 las identificadas, pero la gran mayoría –hasta el 95%– no tienen ni cura ni tratamiento farmacológico específico. Los pacientes pueden tardar más de diez años en tener un diagnóstico certero. Y, cuando llega, el impacto es brutal. Tanto para la persona que está enferma como para el entorno que la rodea.
Según indica la Federación Española de Enfermedades Raras (FEDER), una enfermedad se considera rara en Europa cuando tiene una incidencia inferior a cinco por cada 10.000 habitantes. Son patologías caracterizadas por una amplia diversidad de síntomas que varían según el paciente, sin poder establecerse con certeza cuáles son aquellos más o menos comunes.
Entre las iniciativas desarrolladas en España para mejorar el conocimiento de estas enfermedades, las convocatorias CaixaResearch de Investigación en Salud y CaixaImpulse de Innovación en Salud de Fundación ”la Caixa” trabajan para arrojar luz sobre un campo en el que "todavía falta mucho por saber". "Son pocos los hospitales preparados para atender a los pacientes afectados por una enfermedad rara. Cuando llega el diagnóstico, la mayoría de las familias deben trasladarse de ciudad para seguir el tratamiento, alejándose así de su red familiar, lo que complica aún más la gestión de toda la situación que están viviendo", explica el doctor Manel del Castillo, director gerente del Hospital Sant Joan de Déu.
Infancias afectadas, vidas marcadas
Aunque una enfermedad rara "le puede ocurrir a cualquiera, en cualquier etapa de la vida", tal y como señalan desde las asociaciones de afectados, los pacientes más vulnerables son los niños. Hace pocas semanas, una desconocida politóloga y activista acudía como invitada a La Revuelta. Su nombre es Noah Higón. Padece siete enfermedades raras. En su aparición televisiva para dar visibilidad a esta situación, recordó que "hay muchos niños y niñas que vienen detrás que se merecen un futuro muchísimo mejor que el que yo he tenido. Un presente mejor. Por todos ellos hay que luchar".
El 80% de los pacientes debuta en edad pediátrica. A las enfermedades raras se les puede atribuir el 35% de las muertes ocurridas en menores que no llegan al año de edad, del 10% entre 1 y 5 años y del 12% entre los 5 y 15 años. Además, la mayoría son crónicas y degenerativas, provocando un alto grado de dependencia que se extiende a lo largo de toda una vida. "Hablamos de patologías muy graves; de hecho, en la actualidad, 1 de cada 3 niños afectados por estas enfermedades no llega a los cinco años de edad", asegura Manel del Castillo, cuyo hospital es el principal referente español en la atención de estas patologías en niños y niñas.
En las enfermedades minoritarias, el desarrollo de proyectos de investigación va más allá de conseguir un resultado clínico. Con ocasión del Día Internacional de las Enfermedades Raras, Fundación ”la Caixa” pone el foco sobre los avances médicos en dos de estas patologías para demostrar que hay esperanza en encontrar tratamientos si se apuesta por la investigación primaria.
Una posible cura para la ataxia de Friedreich
Las ataxias son un grupo muy amplio y heterogéneo de enfermedades neurodegenerativas poco frecuentes que afectan de manera preferente el cerebelo, la médula espinal y los nervios que controlan las contradicciones musculares y, por tanto, el movimiento de las extremidades. Una de las más frecuentes, la ataxia de Friedreich, causada por una alteración genética, provoca neurodegeneración severa progresiva, afectación cardíaca y problemas de movimiento.
Tal y como explica Antoni Matilla, investigador del Instituto de Investigación Germans Trias i Pujol (IGTP) y cofundador de la empresa Biointaxis S.L., "la persona afectada empieza a notar falta de sensibilidad en las piernas, tiene dificultades para caminar, tropieza, se cae y es entonces cuando acude al neurólogo. Ante esta descoordinación motora, el neurólogo puede solicitar un diagnóstico genético para confirmar las sospechas de ataxia".


La ataxia de Friedreich está causada por una mutación del gen FXN, que se encuentra en el cromosoma 9. Esta mutación conduce a una deficiencia en la producción de una proteína llamada frataxina, que es esencial para la función normal de las células. Esta deficiencia, a su vez, provoca una acumulación alta y tóxica de hierro dentro de las células que ocasiona daño celular y disfunción mitocondrial.
Por ello, desde hace más de 10 años, el equipo de Matilla trabaja para diseñar secuencias de ADN virales que sustituyan a las dañadas. "Hemos diseñado una estrategia para reemplazar los niveles de la proteína frataxina utilizando un virus ADN asociado recombinante al que le hemos sacado todas las secuencias que podrían ser tóxicas o anómalas para poder así distribuir la proteína que nos interesa", explica. Se espera que el fármaco se empiece a tratar en ensayos clínicos con humanos a partir de 2026. "Con los resultados que tenemos hasta ahora, hablamos de una terapia totalmente curativa. Por un lado, podemos prevenir que los pacientes desarrollen la enfermedad, y por otro, en aquellos que ya la tienen, mejoraríamos el manejo de los síntomas y, por ende, de la calidad de vida", añade el experto.
La enfermedad de Alexander, clave para entender el Alzheimer
La enfermedad de Alexander es una enfermedad rara incluida dentro de las leucodistrofias, caracterizadas por la destrucción de las vainas de mielina que protegen las fibras nerviosas y permiten que los impulsos nerviosos se transmitan de forma rápida y normal. Este trastorno se debe a mutaciones en la proteína ácida fibrilar glial (GFAP), que se encuentra en los astrocitos, un tipo de células esenciales para el correcto funcionamiento del sistema nervioso central.
Los síntomas varían según el tipo de mutación y la edad del paciente, pero algunos permiten reconocer rápidamente la afección, como retrasos en el desarrollo en bebés y niños, convulsiones, ataxia, dificultades en el habla y el lenguaje e incluso macrocefalia. "En adultos pueden aparecer síntomas leves o inespecíficos que se confunden con los de otras enfermedades neurodegenerativas", explica la investigadora Dolores Pérez-Sala, líder del equipo del Centro de Investigaciones Biológicas Margarita Salas del CSIC. Los avances recientes permiten que la enfermedad de Alexander pueda diagnosticarse en trastornos cuya causa era antes desconocida.

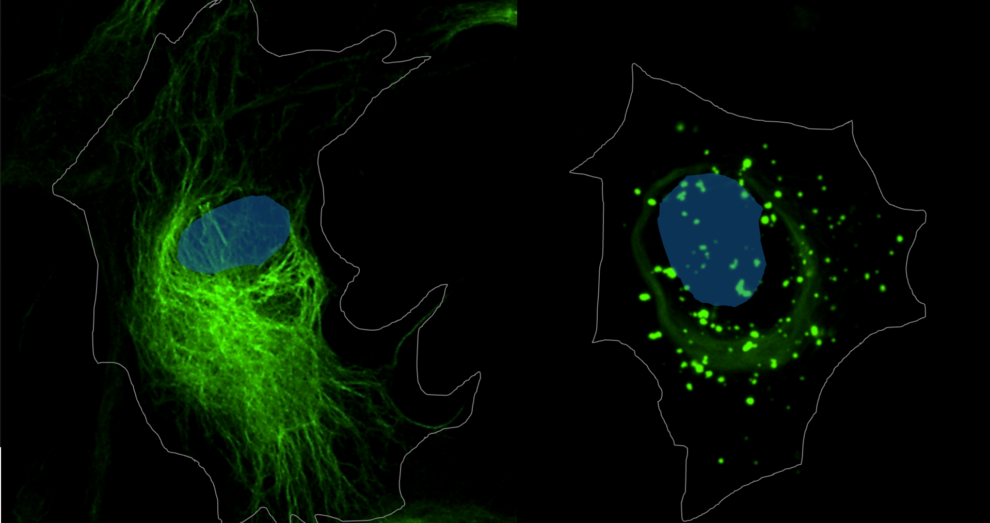
Aunque el tratamiento actual de esta enfermedad es fundamentalmente sintomático y varía según el paciente, el equipo de Pérez-Sala está desarrollando un ensayo clínico con un fármaco experimental diseñado para inhibir la producción de GFAP. "El objetivo es reducir los niveles de la proteína anómala para evitar que deteriore tanto la función de la célula. Se espera conocer los resultados, al menos sobre la seguridad del fármaco, hacia finales de este año", desvela la experta.
El estudio ha arrojado también ciertos destellos de luz que permiten comprender los mecanismos de otras enfermedades neurodegenerativas no tan extrañas. "La enfermedad de Alexander comparte diversas características con enfermedades como el Alzheimer o la esclerosis lateral amiotrófica (ELA) e incluso con los problemas derivados de traumatismos o accidentes cerebrovasculares. Por eso esperamos que nuestros hallazgos también puedan ser útiles en la lucha contra esas enfermedades", prevé Pérez-Sala.
El desafío de la financiación
Las enfermedades raras son un reto para el paciente, pero también para los expertos. Matilla asegura que el principal desafío al que se enfrentan a la hora de avanzar en el diagnóstico y tratamiento de enfermedades minoritarias es la financiación. "Afortunadamente, cada vez hay más empresas farmacéuticas que apuestan por desarrollar fármacos y terapias contra las enfermedades raras, pero la financiación no está al nivel de otras patologías más comunes, como el VIH o el cáncer", incide.
Dolores Pérez-Sala reconoce un desafío adicional, relacionado con la mínima incidencia de estas enfermedades entre la población. "Es complicado desarrollar modelos experimentales adecuados, sobre todo en casos en los que no se conoce la causa de la enfermedad o se trata de enfermedades multifactoriales, lo que implica un reto adicional a la hora de encontrar posibles tratamientos", explica. Por su parte, Manel del Castillo incide en que, para un mejor futuro en la investigación de estas patologías, "hemos de conseguir centros integrados, que den una atención más global a los pacientes; más traslacionales, que sean capaces de trabajar en la asistencia y la investigación de forma conjunta; y más cooperativos, que incorporen a la búsqueda de soluciones al conjunto de la sociedad, a otros centros de investigación y a las familias
En los últimos diez años, la Fundación ”la Caixa” ha apoyado más de 45 proyectos de investigación e innovación en enfermedades raras. Además, recientemente ha destinado 7,5 millones de euros a impulsar la investigación del Hospital Sant Joan de Déu en enfermedades minoritarias pediátricas, con un doble objetivo: mejorar el diagnóstico con técnicas de aprendizaje automático y facilitar la investigación de nuevos tratamientos.
Te puede interesar
Lo más visto
- 1 Otra actriz de La Promesa se marcha y defiende a sus guionistas
- 2 Una actriz de Sueños de libertad anuncia su embarazo
- 3 Trump levanta una muralla a Europa y China, pero exime del castigo a Rusia
- 4 La Fiscalía pide seis años y medio de cárcel para el teniente coronel Oliva
- 5 Madrid se revuelve ante las dudas del Gobierno sobre menores inmigrantes
- 6 Peinado rechaza la petición de Bolaños y le informa de que no puede declarar como testigo por escrito
- 7 Trump impondrá aranceles del 20% para todos los productos de la UE
- 8 Luis García Montero y las viudas vituperadas
- 9 El creador de Adolescencia, serie de Netflix, desmonta su mentira